



- Wissenschaft
- Originaltext
Dipl.-Ing. (FH) Claudia Pachl
Zwischen GMP und Kinderwunsch
Wie Reinraumtechnik in der Assistierten Reproduktionstechnik sinnvoll umgesetzt werden kann
")
Abbildung 5: Embryolabor mit ICSI- und Transferarbeitsplatz (Quelle: Praxis für Fertilität Dr. Peet und Dr. Wilkening)
")
Abbildung 4: Eizelllabor (Quelle: Praxis für Fertilität Dr. Peet und Dr. Wilkening)
 sowie IVF-Werkbänke (Quelle: ZERM)")
Abbildung 6: ICSI-Arbeitsplätze (als TAV-Bereich) sowie IVF-Werkbänke (Quelle: ZERM)
")
Abbildung 7: IVF-Werkbänke (Quelle: ZERM)
")
Abbildung 3: Andrologielabor (Quelle: Praxis für Fertilität Dr. Peet und Dr. Wilkening)
")
Abbildung 8: Transferbereich mit Durchreiche (Quelle: ZERM)
")
Abbildung 9: Andrologielabor (Quelle: ZERM)
")
Abbildung 10: Materialaufzug Punktat (Quelle: ZERM)
")
Abbildung 11: Materialschleuse (Quelle: ZERM)
")
Abbildung 12: Kryolager (Quelle: ZERM)
 Claudia Pachl, VALTEC")
Dipl.-Ing. (FH) Claudia Pachl, VALTEC
Reinraumtechnik im Bereich der Assistierten Reproduktionstechnik (ART) ist ein viel diskutiertes Thema. Im Arzneimittelgesetz (AMG) bzw. der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) geregelt, aber doch nicht vergleichbar mit sonstigen Geweben oder gar der Arzneimittelherstellung, bleibt der Anwender – die Kinderwunschzentren und Arztpraxen – oftmals ratlos zurück. Reinraumtechnik und Kinderwunsch, auf den ersten Blick nur schwer miteinander vereinbar. Doch es gibt verschiedene Optionen und Spielräume, die bei kluger Nutzung zu einem wertschöpfenden und belastbaren System für Anwender und Patientinnen führen können.
Erstveröffentlichung: cleanroom & processes 4, Nr. 3, 2–10 (2025)
Reinraumtechnik findet Anwendung, wo luftgetragene Partikel einen i. d. R. schädlichen Einfluss auf das herzustellende Produkt und die damit verbundenen Herstellprozesse bedeuten. Halbleiterfertigung, Optik- und Lasertechnologie, Luft- und Raumfahrttechnik – nicht möglich ohne Reinräume und Reinraumtechnik. Aber auch in der Lebensmittelindustrie, Biotechnologie, bei Medizinprodukten und Arzneimitteln sind reine und kontrollierte Umgebungsbedingungen grundlegend – abhängig von Herstellverfahren, Produktanforderungen und Anwendung. Neben den luftgetragenen Partikeln spielt hier dann auch noch u. a. die Keimzahl eine wesentliche Rolle.
Ohne Reinraumtechnik kein Smartphone, kein haltbarer Käseaufschnitt und keine (im Notfall lebensrettenden) Medikamente, um nur einige Beispiele zu nennen. Neben dem Schutz des Anwenders – Kunde bzw. Patient – sowie des Produkts selbst, fungiert Reinraumtechnik auch als Schutz der herstellenden Personen sowie der Umwelt.
Ein weiterer Bereich, für den die Anwendung von Reinraumtechnik in den vergangenen Jahren zunehmend in den Fokus gerückt ist, sind die Assistierten Reproduktionstechniken (ART), oftmals vereinfacht als künstliche Befruchtung bezeichnet. Da sich dieser Beitrag nicht detailliert mit den unterschiedlichen Techniken und Methoden im großen Feld der ART beschäftigen soll – z. B. In-Vitro-Fertilisation (IVF), Intrauterine Insemination (IUI) oder Intrazytoplasmatische Spermieninjektion (ICSI) – wird nachfolgend übergreifend der Begriff ART verwendet, unabhängig davon, welche Methode angewendet wird. Fokus in diesem Beitrag ist die Betrachtung, inwieweit die Anwendung von Reinraumtechnik im ART-Bereich einen Nutzen bietet oder eher schädlich ist. Denn genau das wird von Anwendern, Experten und Behörden teils kontrovers diskutiert.
Rechtliche Grundlagen
Im ART-Bereich ist eine Vielzahl an Gesetzen und Verordnungen zu berücksichtigen:
Das Arzneimittelgesetz
Kinderwunschpraxen und Fertilitätszentren benötigen eine Erlaubnis der zuständigen Landesbehörde gemäß §§ 20 b und c des Arzneimittelgesetzes (AMG) [1], um Aktivitäten im Rahmen einer Kinderwunschbehandlung durchführen zu dürfen. Der § 20 b regelt die Gewinnung von Gewebe und die Laboruntersuchungen, § 20c behandelt die Be- oder Verarbeitung, Konservierung, Prüfung, Lagerung sowie das Inverkehrbringen von Gewebe oder Gewebezubereitungen.
Es finden sich hier auch Angaben zur Sachkundigen Person und deren erforderlicher Qualifikation, die Forderung nach qualifiziertem Personal sowie nach geeigneten Räumen und Einrichtungen für die beabsichtigten Tätigkeiten. Außerdem enthält das AMG auch den Hinweis auf die erforderliche Anwendung der Guten Fachlichen Praxis (GFP) sowie die Berücksichtigung des aktuellen Stands von Wissenschaft und Technik bei der Be- oder Verarbeitung, einschließlich der Kennzeichnung, Konservierung und Lagerung sowie der Prüfung. Und zuletzt findet sich der Verweis auf ein Qualitätsmanagementsystem (QMS) nach den Grundsätzen der GFP, das kontinuierlich zu pflegen ist.
Arzneimittel- und Wirkstoffherstellungsverordnung
In der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) regelt Abschnitt 5a [2] die Sondervorschriften für Entnahme- und Gewebeeinrichtungen sowie für Gewebespenderlabore. Mit Blick auf den Fokus dieses Beitrags ist besonders § 36 „Be- oder Verarbeitung und Lagerung durch die Gewebeeinrichtung“ interessant. Dort heißt es in Abs. (2):
„Die Betriebsräume und Ausrüstungen nach § 5 und die Hygienemaßnahmen nach § 6 müssen geeignet sein, die Eigenschaften des Gewebes zu schützen, die für seine Verwendung erforderlich sind, und das Risiko einer Verunreinigung, insbesondere einer mikrobiellen Verunreinigung, während der Be- oder Verarbeitung zu minimieren:
Soweit die Gewebe während ihrer Be- oder Verarbeitung der Umgebung ausgesetzt werden, muss dies in einer Umgebung mit festgelegter Luftqualität und Sauberkeit erfolgen. Die Wirksamkeit dieser Maßnahmen ist zu validieren und zu überwachen.
Sofern die Gewebe nach Nummer 1 keinem Inaktivierungs- oder Sterilisationsverfahren unterzogen werden, ist während der Be- oder Verarbeitung ein Luftreinheitsgrad für Keimzahl und Partikelzahl entsprechend Klasse A der Definition des EU-GMP-Leitfadens, Anhang 1 (Bekanntmachung vom 12. März 2008, BAnz. S. 1217), mit einer für die Be- oder Verarbeitung des Gewebes geeigneten Hintergrundumgebung, die in Bezug auf Partikel- und Keimzahl mindestens der Klasse D des Anhangs 1 des Leitfadens entspricht, erforderlich.“ [2]
Hier findet sich also ganz klar die Forderung nach einer Reinheitsklasse A mit einer Hintergrundumgebung von mindestens D gemäß EU-GMP-Leitfaden [3] bzgl. der Partikel und Keime. Folgende Ausnahmen werden eingeräumt:
„Von den Anforderungen an die Umgebung kann abgewichen werden, wenn
a) ein validiertes Verfahren zur Inaktivierung der Keime oder zur Endsterilisation angewendet wird oder
b) nachgewiesen wird, dass die Exposition gegenüber einer Umgebung der Klasse A schädliche Auswirkungen auf die erforderlichen Eigenschaften der Gewebe hat, oder
c) nachgewiesen wird, dass mit der Art und Weise der Verwendung der Gewebe beim Empfänger oder bei der Empfängerin ein erheblich geringeres Risiko der Übertragung einer bakteriellen oder Pilzinfektion auf den Empfänger oder die Empfängerin einhergeht als bei der Gewebetransplantation, oder
d) es technisch nicht möglich ist, das erforderliche Verfahren in einer Umgebung der Klasse A durchzuführen.
Es ist nachzuweisen und zu dokumentieren, dass mit der gewählten Umgebung die erforderliche Qualität und Sicherheit des Gewebes oder der Gewebezubereitung erreicht wird, mindestens unter Berücksichtigung des Bestimmungszwecks, der Art der Verwendung und des Immunstatus des Empfängers oder der Empfängerin.“ [2]
Da die Keimzellen im Bereich der Assistierten Reproduktionstechniken selbstverständlich keinem Inaktivierungs- oder Sterilisationsverfahren unterzogen werden können, sind somit die grundlegenden Anforderungen im Hinblick auf die Umgebungsbedingungen eindeutig definiert. Die in [2] unter a) beschriebene mögliche Abweichung von diesen Anforderungen kommt nicht zur Anwendung. Für die unter c) beschriebene Option existieren bereits verschiedene Fachpublikationen, Statements der einschlägigen Fachkreise sowie Risikobewertungen. Der Transfer von Keimzellen und Embryonen auf die Patientin birgt zwar verschiedene Verletzungsrisiken, unterscheidet sich aber grundlegend von den Risiken, die mit einer Gewebetransplantation verbunden sind.
Da der Fokus dieses Beitrags auf den Einsatz von Reinraumtechnik im ART-Bereich gelegt werden soll und nicht auf medizinische Detailaspekte, wird hier auf eine vertiefte Darstellung verzichtet und auf die einschlägige Literatur und Fachpublikationen verwiesen.
Die Optionen b) und d) sind wiederum direkt mit der Anwendung von Reinraumtechnik verbunden und werden in diesem Beitrag näher betrachtet.
EU-GMP-Leitfaden
Der Annex 1 des EU-GMP-Leitfadens [3] ist gemäß Referenz in Abschnitt 5a, § 36 ff der AMWHV maßgebend im Hinblick auf die Partikel und Keime. Es gelten die gemäß Annex 1 definierten Grenzwerte, wobei in der AMWHV noch der alte Annex 1 vom 12. März 2008 referenziert ist. Da die Grenzwerte bzgl. Partikel und Keime jedoch identisch sind mit der aktuellen Fassung vom 22. Aug .2022, stellt dies für die Betrachtung in diesem Beitrag kein Problem dar.
Die übrigen Parameter gemäß EU-GMP-Leitfaden, die im Annex 1 für die Klassen A–D beschrieben sind, werden in der AMWHV Abschnitt 5a, § 36 ff nicht erwähnt und sind damit für den ART-Bereich auch nicht explizit gefordert, z. B. Strömungsgeschwindigkeit, Luftwechsel, Differenzdrücke usw.
Der EU-GMP-Leitfaden ist gemäß GFP-Leitfaden der Deutschen Gesellschaft für Chirurgie [4] nicht anwendbar für die Gewinnung von menschlichen Geweben und Zellen (gemäß § 20 b AMG), sofern die Entnahme nach den GFP-Regeln erfolgt.
Arbeitsgemeinschaft Reproduktionsbiologie des Menschen
Die Arbeitsgemeinschaft Reproduktionsbiologie des Menschen (AGRBM) hat 2 Leitfäden publiziert, die sich mit der Führung und Einrichtung eines ART-Labors [5] sowie dem verantwortlichen Arbeiten im ART-Labor [6] beschäftigen.
Diese Leitlinien behandeln ebenfalls die Frage nach den Umgebungsbedingungen nach Annex 1 EU-GMP-Leitfaden und den Auswirkungen auf die Keimzellen. Das ART-Labor ist gemäß AGRBM-Leitlinie Teil einer Entnahmeeinrichtung.
Nach Auffassung der AGRBM und der European Society of Human Reproduction and Embryology (ESHRE) [7] sind die Ausnahmeregelungen, die in der AMWHV Abschnitt 5a in den Punkten a) bis d) genannt werden, vollumfänglich auf den ART-Bereich und die darin ablaufenden Prozesse anzuwenden. Umgebungsbedingungen einer Reinheitsklasse D nach Annex 1 werden trotzdem empfohlen, Klasse A Bedingungen werden jedoch als kontraproduktiv bewertet und nicht empfohlen.
Sonstige Regularien und Vorschriften
Neben den bereits genannten gibt es noch zahlreiche weitere Regularien und Anforderungsdokumente, die für den ART-Bereich anwendbar sind. Hier seien vor allem erwähnt das Transplantationsgesetz [8], das Embryonenschutzgesetz [9], das Infektionsschutzgesetz [10] und die ESHRE-Guidelines [11]. Ergänzt wird dies durch den European Directorate for the Quality of Medicines & HealthCare (EDQM) Guide [12] sowie der neuen Substances-of-Human Origin(SoHO)-Verordnung [13], die ab 2027 gültig sein wird.
Bezüglich Reinraumtechnik sind u. a. die entsprechenden Teile der technischen Regeln gemäß DIN EN 14644 [14] sowie VDI 2083 [15] zu berücksichtigen.
Patientensicherheit
Auch ohne tieferes medizinisches Wissen ist nachvollziehbar, dass sich eine Transplantation von Stammzellen oder Organen von einer künstlichen Befruchtung bzw. einem Embryotransfer unterscheidet. Im ersten Fall handelt es sich i. d. R. um schwer kranke, teils immunsupprimierte Patienten, deren Überleben mehr oder weniger von der Gewebetransplantation abhängt. Das transplantierte Gewebe ist z. T. als steril spezifiziert, Gewinnung, Be- und Verarbeitung erfolgen i. d. R. aseptisch bzw. unter weitestgehend aseptischen Bedingungen. Die Prozessschritte und das Equipment sind entsprechend darauf angepasst und laufen zu einem großen Teil in geschlossenen Systemen ab. Die Transplantation findet im Normalfall unter OP-Bedingungen statt.
Im Fall der ART sind die Empfängerinnen i. d. R. gesund bzw. haben einen guten Immunstatus, auch wenn es vielfältige Gründe gibt, die ursächlich für den unerfüllten Kinderwunsch sein können. Die Gewinnung der Keimzellen sowohl bei der Frau als auch beim Mann kann nicht keimfrei erfolgen. Die Rückführung der bearbeiteten Zellen in den Uterus der immunkompetenten Empfängerin erfolgt über eine natürliche Körperöffnung, die natürlicherweise mit Keimen besiedelt ist. Sollte es während der extrakorporalen Kultivierung zu einer von außen eingetragenen Kontamination mit Keimen kommen, wird die Kultivierung bzw. der Transfer schlimmstenfalls ausgesetzt. Die Verletzungsgefahr bei Gewinnung und Rückführung ist gering und nicht vergleichbar mit einer Transplantation.
Insofern darf infrage gestellt werden, ob die vollumfängliche Anwendung einer Reinheitsklasse A für kritische Schritte mit einer Hintergrundumgebung von mindestens Klasse D zwingend erforderlich ist oder ob die Sicherheit der Keimzellen und Embryonen durch keimreduzierte bzw. keimarme Prozessabläufe im ART-Bereich bei der Be- und Verarbeitung und damit letztendlich die Patientensicherheit nicht auch anders gewährleistet werden kann.
Reinraumtechnik für ART – sinnvoll oder schädlich?
Wie bereits erwähnt, geben die Regularien nur auf den ersten Blick eindeutige Vorgaben bzgl. der Umgebungsbedingungen, da die relevanten Passagen auf Gewebe im Allgemeinen bezogen sind und nicht auf den ART-Bereich im Speziellen. Die Gewinnung, Be- und Verarbeitung von Geweben sowie die Übertragung auf die Patienten ist jedoch so vielfältig und je nach Anwendungsfall speziell, dass der regulatorische Versuch, allen Varianten mit einem gemeinsamen Passus gerecht zu werden, aus Sicht der Autorin etwas unglücklich ist und folglich für Kontroversen sorgt.
Für die Be- bzw. Verarbeitung von Keimzellen im ART-Bereich, die unter § 20 c AMG fällt, erwarten die Inspektoren der Landesbehörden kontrollierte Umgebungsbedingungen, wie in der AMWHV, Abschnitt 5a gefordert. Darüber hinaus die Qualifizierung von Ausrüstung und Gerätschaften, die Überwachung kritischer Parameter sowie ein QMS entsprechend der GFP. Das führt erfahrungsgemäß regelmäßig zu Diskussionen und auch zu Verunsicherung auf Seiten der Anwender.
Die Fachkreise betonen die uneingeschränkte Anwendbarkeit der in der AMWHV beschriebenen Ausnahmeregelungen (siehe Abschnitt Arzneimittel- und Wirkstoffherstellungsverordnung) und betonen den schädlichen Einfluss von Reinraumtechnik und den damit verbundenen technischen Maßnahmen und geforderten Umgebungsbedingungen. In der AGRBM-Leitlinie [5] werden die Ausnahmeregelungen ausführlich betrachtet und begründet.
So wird vor allem der negative Einfluss der Strömungsgeschwindigkeit in Bereichen der GMP-Klasse A hervorgehoben, der zu Abkühleffekten, Austrocknung, Änderung des extrazellulären pH-Werts sowie Änderung der Osmolarität im Medium führt und damit letztendlich die Zellen schädigt. Zudem wird durch die zur Einhaltung von klassischen GMP-Bedingungen erforderliche Technik sowie Hygienemaßnahmen das Handling mit den Keimzellen deutlich erschwert, was zu einem hohen Risiko für das Produkt führt und eine Gefahr für die Keimzellen bedeutet. Zum Teil können klassische GMP-Anforderungen v. a. für eine Klasse A im ART-Bereich zudem technisch nicht umgesetzt werden. So ist die Platzierung eines ICSI-Mikroskops mit Zubehör in einer Werkbank technisch fast nicht machbar und die Handhabung bzw. die korrekte Durchführung der entsprechenden Prozessschritte nahezu unmöglich.
Die inspizierenden Behördenvertreter sind ihrerseits wiederum an die anzuwendenden Regularien gebunden und fordern demzufolge entsprechende Nachweise und Dokumente, mit denen die Einhaltung der regulatorischen Anforderungen nachgewiesen wird. Stellungnahmen von Arbeitsgruppen und Fachkreisen finden da wenig Berücksichtigung. Eine herausfordernde Situation, die mit ein wenig Kreativität aber durchaus zu lösen ist.
Lösungsansätze
Die Autorin vermutet, dass höchstwahrscheinlich aus Mangel an Alternativen die AMWHV in Abschnitt 5a auf den Annex 1 des EU-GMP-Leitfadens [3] verweist bzgl. der Definition von Partikeln und Keimen.
Bei genauer Betrachtung beschränkt sich diese Angabe nämlich ausschließlich auf diese. Sämtliche anderen relevanten Parameter, die im Annex 1 beschrieben sind, werden nicht erwähnt oder gefordert, z. B. Differenzdrücke, Luftwechsel, Erholzeit. Und auch die Strömungsgeschwindigkeit, die in einer turbulenzarmen Verdrängungsströmung für eine Klasse A mit 0,36 m/s bis 0,52 m/s als Stand der Technik implementiert ist, wird in den anzuwendenden Abschnitten für Gewebe – und damit auch für den ART-Bereich – nirgends explizit gefordert. Das lässt Raum für Interpretation und kreative Auslegung und bietet damit verschiedene Möglichkeiten für die technische Umsetzung, die in der klassischen Pharmafertigung oder der Medizintechnik gar nicht diskussionsfähig wären.
Abgesehen davon ist der EU-GMP-Leitfaden mit seinen Annexen ursprünglich als Leitlinie für die Herstellung von Arzneimitteln für Mensch und Tier geschaffen worden, die üblicherweise unter definierten Bedingungen, mit standardisierten Abläufen und reproduzierbaren Ergebnissen abläuft. Der Annex 1 des Leitfadens hat explizit die Herstellung steriler Arzneimittel im Fokus.
Die Prozesse im ART-Bereich folgen zwar ebenfalls – methodenabhängig – definierten und etablierten Abläufen. Trotzdem sind selbst bei identischen Umgebungsbedingungen und Ablaufstrukturen patientenindividuell höchst unterschiedliche Ergebnisse zu erwarten. So können die Eizellen von ein und derselben Spenderin unterschiedliche Qualitäten besitzen, sich unter identischen Umgebungsbedingungen und unter Verwendung derselben Medien unterschiedlich entwickeln und für die weitere Verwendung besser oder weniger gut geeignet sein. Und keinesfalls ist das Endprodukt steril.
Aus Sicht der Autorin kann der Annex 1 des EU-GMP-Leitfadens somit als Orientierungshilfe verstanden werden, um die Anforderungen hinsichtlich Partikel- und Keimzahlen zu erreichen. Die vollumfängliche Umsetzung der sonstigen Anforderungen gemäß Annex 1 ist für den ART-Bereich jedoch nirgends gefordert. Gefordert sind also die Partikel- und Keimzahlen nach Annex 1, aber ohne den nach aktuellem Stand der (Reinraum-)Technik dafür erforderlichen technischen Aufwand.
Wie kann das in der Praxis nun aussehen? Ganz ohne technisches Equipment und Installationen geht es leider nicht. Und um Partikel und Keime einer Klasse D bzw. A sicherstellen zu können, braucht es zwangsläufig auch gewisse Rahmenbedingungen. Die Luft muss entsprechend geführt und aufbereitet werden, kritische Parameter sind zu überwachen, Oberflächen müssen geeignet sein im Hinblick auf Partikelabgabe, flüchtige organische Verbindungen (Volatile Organic Compounds, VOC) sowie Beständigkeit gegenüber den einzusetzenden Reinigungs- und Desinfektionsmitteln.
Das alles bedingt auch gewisse bauliche Voraussetzungen, um nicht an anderen Stellen z. B. Schwierigkeiten mit der Arbeitssicherheit zu bekommen (z. B. bei den Raumhöhen, wenn Kanäle an der Decke geführt werden). Das macht technisch notwendige Installationen in Gebäuden, die als Praxis- oder Büroräume konzipiert wurden, einigermaßen herausfordernd bis z. T. unmöglich.
Volatile Organic Compounds
An dieser Stelle sei auf einen weiteren wesentlichen Aspekt hingewiesen, der in den Regularien der inspizierenden Behörden nirgends erwähnt wird: VOC. Es handelt sich hierbei um gas- und dampfförmige Stoffe organischen Ursprungs in der Luft, z. B. Kohlenwasserstoffe, Alkohole, Aldehyde und organische Säuren. Diese können entweder aus den verwendeten Materialien von Oberflächen (Decken, Böden, Wände, Mobiliar usw.) austreten oder während der Reinigung und Desinfektion entstehen.
VOC sind besonders schädlich für die Keimzellen im ART-Bereich und sollten deshalb weitestgehend vermieden bzw. reduziert werden. Sie haben einen zytotoxischen Effekt und können die Qualität der Keimzellen bzw. deren Entwicklung negativ beeinflussen. Zahlreiche Publikationen der einschlägigen Fachkreise belegen dies.
Paradoxerweise wird dieser Aspekt in Behördeninspektionen so gut wie nie berücksichtigt, da die entsprechenden Vorgaben und Hinweise in den anzuwendenden Passagen für die Erlaubnis nach AMG § 20 b und c [1] nicht enthalten ist. Bei der Planung und Gestaltung von Räumlichkeiten und Umgebungsbedingungen im ART-Bereich muss dies jedoch zwingend berücksichtigt werden. Für die Reinigung und Desinfektion stellt dies eine besondere Herausforderung dar, um die im GMP-regulierten Umfeld etablierten Anforderungen für die Keimzellen kompatibel umzusetzen.
Der negative Einfluss von VOC auf die Keimzellen ist ungleich größer als eine potenzielle Kontamination durch Partikel und Keime. Hilfestellung zum Thema VOC liefern z. B. DIN EN ISO 14644-8 Klassifizierung der Luftreinheit anhand der Chemikalienkonzentration [15] sowie VDI 2083-8.1 Luftreinheit anhand chemischer Konzentration (ACC) [16]. Konkrete Grenzwerte für den ART-Bereich sucht man jedoch in der Literatur vergeblich.
Praxisbeispiele
Nachfolgend zeigen 2 Projektbeispiele, wie die Compliance zwischen den regulatorischen Vorgaben zur Erreichung der Erlaubnis nach AMG § 20 b und c, den Anforderungen gemäß der GFP und den Fachgremien, sowie den prozessspezifischen, Betreiber-individuellen Bedingungen hergestellt werden kann.
Fall 1:
Praxis für Fertilität Dr. Peet und Dr. Wilkening in Berlin, in einem Gebäudekomplex mit Hotel und sonstigem Gewerbe. Umzug der Praxis in die neuen Räume im Jahr 2016. Ausgangssituation (Abb. 1):
– keine klare Abgrenzung des ART-Bereichs
– kein abgestimmtes Personal- und Materialflusskonzept
– keine kontrollierten Umgebungsbedingungen
– keine Qualifizierung der Geräte und Umgebung

Abbildung 1: Layout des ART-Bereichs. (Quelle: Praxis für Fertilität Dr. Peet und Dr. Wilkening)
Die zuständige Behörde hat die Erlaubnis nach AMG § 20 b und c verweigert. Der Ansatz sowie die Prozessabläufe und Umgebungsbedingungen waren analog den vorher bestehenden Räumen in derselben Straße. Durch den Umzug in die neuen Räume erlosch der Bestandsschutz und es mussten vollumfänglich die Anforderungen des AMG [1] bzw. der AMWHV [2] erfüllt werden.
Umsetzung:
Das Layout wurde bezüglich der Raumgeometrie nicht verändert. Der Laborbereich wurde jedoch als Reinraumbereich klar von den übrigen Praxisbereichen abgetrennt, das bestehende Zutrittskonzept entsprechend angepasst. Es wurden Reinraumklassen in Anlehnung an den Annex 1 des EU GMP-Leitfadens definiert, die Räume und Verkehrsflächen entsprechen überwiegend einer Klasse D (Abb. 2, in rot dargestellt). Der offene Umgang mit den Keimzellen erfolgt unter Klasse A – Bedingungen (Abb. 2, in blau dargestellt), die vor allem über Bereiche mit turbulenzarmer Verdrängungsströmung (TAV) hergestellt werden. Besonders ist, dass es sich hierbei um ein offenes A in D – Konzept handelt. Dies war notwendig, um die ART-spezifischen Prozessabläufe mit möglichst wenig Einschränkungen bei gleichzeitig maximaler Keimreduktion sicherzustellen (Abb. 3, 4 und 5). Trotz der prozessrelevanten Ausrüstungsgegenstände im A-Bereich – z.B. Zentrifugen, Inkubatoren, Mikroskope mit Wärmeplatten – wird eine turbulenzarme Verdrängungsströmung bei reduzierter Strömungsgeschwindigkeit erreicht. Dies wird durch regelmäßige Messungen zum Nachweis des qualifizierten Zustands nach DIN EN ISO 14644 [14] überprüft und belegt.

Abbildung 2: GMP-konformes Layout des ART-Bereichs. (Quelle: Praxis für Fertilität Dr. Peet und Dr. Wilkening)
Fall 2:
IVF-Labor im Zentrum für Endokrinologie und Reproduktionsmedizin (ZERM) am Universitätsklinikum Würzburg
Ausgangssituation:
– aktuelle Räumlichkeiten waren veraltet und zu klein
– Umbau im Turm auf 3 Etagen, IVF-Labor im 3. Stock
– Bestandsgebäude wurde entkernt und neu geplant
Umsetzung:
In diesem Projekt konnte in dem entkernten Stockwerk ein vollständig neues Layoutkonzept für den Laborbereich erarbeitet werden. Auch hier wurde ein Klasse A in D - Konzept zu Grunde gelegt. Aufgrund kundenspezifischer Vorgaben werden die Klasse A – Bereiche entweder über IVF-Werkbänke (Abb. 6, 7 und 9) sowie einen großen TAV-Bereich mit zwei ICSI-Arbeitsplätzen (Abb. 6) realisiert.
Eine besondere Herausforderung in diesem Projekt war der Transport des Punktats (Eizellen) aus dem 1. OG in das IVF-Labor im 3. OG. Hierfür wurde ein GMP-gerechter Materialaufzug (Abb. 10) konstruiert, der im Innenraum Bedingungen der Klasse D gewährleistet und zudem die geforderte Druckkaskade bzw. Strömungsrichtung vom IVF-Labor zum Entnahmeraum über zwei Geschosse sicherstellt.
Darüber hinaus wurde eine Durchreiche (Abb. 8) neben dem Andrologielabor zum angrenzenden Transferraum (außerhalb des Reinraumbereichs) installiert, über die der Transferkatheter aus dem IVF-Labor in den Transferraum und anschließend wieder zurück gereicht wird. Das Kryolager (Abb. 12) ist über den Schwarzbereich der Materialschleuse zugänglich und darüber in das allgemeine Zutrittskonzept eingebunden. Es unterliegt als CNC-Bereich (controlled, not classified) weniger strengen Umgebungsanforderungen, fügt sich jedoch optimal in den Prozessflow und das Hygienekonzept ein.
Lösungsansätze in beiden Projekten:
– Überprüfung und Anpassung der Prozesse und Abläufe an die neuen Gegebenheiten
– risikobasierte Identifikation der kritischen Prozessschritte und Parameter
– klare Abgrenzung des reinen Bereiches, inkl. Zutrittskonzept
– Absicherung der kritischen Prozessschritte und Parameter (technisch, organisatorisch)
– Schaffung bestmöglicher Umgebungsbedingungen ohne negative Beeinflussung der Keimzellen
– optimale Anordnung der Bereiche und Installationen
– Installation RLT-Anlage und von Bereichen mit turbulenzarmer Verdrängungsströmung (TAV) (Abb. 6) sowie IVF-Werkbänke (Abb. 7, Abb. 9) mit jeweils angepasster Parametrierung
– Anpassung des Hygienekonzepts (Bekleidung, Reinigung und Desinfektion)
– Qualifizierung aller relevanten Geräte, Umgebungsbedingungen, Lager
– Wirksamkeitsnachweis Reinigung und Desinfektion
– QMS, inkl. Standard Operating Procedures (SOPs)
Durch gezielt ausgewählte technische Maßnahmen, wie z. B. RLT-Anlagen, den Prozessabläufen entsprechend dimensionierte TAV-Bereiche, IVF-Werkbänke (wo erforderlich), Druckstufenkonzept, geeignete Oberflächen und Materialien (Stichwort: VOC) sowie einem nutzergerechten Hygiene- und Bekleidungskonzept wurden bestmögliche Bedingungen geschaffen, die ein sicheres Handling der Keimzellen mit möglichst geringer Beeinflussung der Arbeitsabläufe durch die Reinraumtechnik ermöglichen. Vor allem die Schaffung großzügiger TAV-Bereiche als offener Klasse-A-Bereich in einer Klasse-D-Umgebung ermöglichen meist ein deutlich besseres Handling bei den betreffenden Prozessschritten, als dies in einer Werkbank möglich wäre. Natürlich setzt dies entsprechende gebäudetechnische Gegebenheiten voraus.
Ergänzt wurde das Konzept jeweils durch eine anwendungsgerechte und sinnvolle Parametrierung, z. B. wurde die Strömungsgeschwindigkeit in den A-Bereichen deutlich unter 0,36 m/s abgesenkt, um negative Effekte auf die Keimzellen bei offenem Handling möglichst zu vermeiden. Die Aufrechterhaltung einer TAV trotz geringer Strömungsgeschwindigkeit und bei installiertem Equipment im A-Bereich (z. B. Inkubatoren, Mikroskope mit Wärmeplatten, Zentrifugen) wurde mittels Strömungsvisualisierung zuverlässig nachgewiesen.
Das Offene-A-in-D- Konzept stellt die regulatorischen Mindestanforderungen nach AMWHV sicher. Die Grenzwerte für Partikel und Keime nach Annex 1 des EU-GMP-Leitfadens werden sicher eingehalten (Erfassung der Werte in der Praxis für Fertilität seit 2017, im UKW seit Ende 2023). In anderen Life-Science-Bereichen u. U. ein nicht diskussionsfähiger Ansatz, ist er im IVF-Bereich aufgrund der Vorgaben jedoch möglich und durch die Praxistauglichkeit belegt. Dieser Ansatz konnte in einem weiteren Projekt auch bereits im Stammzelllabor eines Spitals erfolgreich eingesetzt werden.
Über ein entsprechend ausgelegtes kontinuierliches Monitoringsystem im ART-Bereich, das risikobasiert an den erforderlichen Positionen installiert und parametriert wurde, ist die permanente Überwachung der kritischen Parameter sichergestellt. Es findet kein produktionsbegleitendes Partikelmonitoring in Klasse A statt, dies konnte über eine entsprechende Risikobetrachtung ausgeschlossen werden. Ein entsprechendes Hygienemonitoring sowie ein diskontinuierliches Monitoring zur Aufrechterhaltung des qualifizierten Zustands vervollständigen das Überwachungskonzept.
Sämtliche relevanten Geräte, Ausrüstungsgegenstände und Umgebungsbedingungen wurden vollumfänglich qualifiziert. Die Wirksamkeit der eingesetzten Reinigungs- und Desinfektionsmittel wurde nachgewiesen.
Grundlage für sämtliche Arbeiten ist ein angepasstes QMS, das für diesen Bereich entsprechend modifiziert wurde. Da die Nutzer in einem IVF-Labor i. d. R. nicht über vergleichbare Ressourcen wie z. B. in der Pharmaindustrie in diesem Bereich verfügen, muss das QMS die regulatorischen Anforderungen sowie die Produkt- und Patientensicherheit berücksichtigen, andererseits aber auch einen leistbaren Aufwand für den Nutzer darstellen, damit das System langfristig gelebt wird.
Statistik
Am Beispiel des IVF-Labors am UKW wurde vom Nutzer folgende Kurzstatistik zur Verfügung gestellt (Tab. 1):
Tabelle 1: Statistik Embryonentransfers (ETs) und Schwangerschaften.
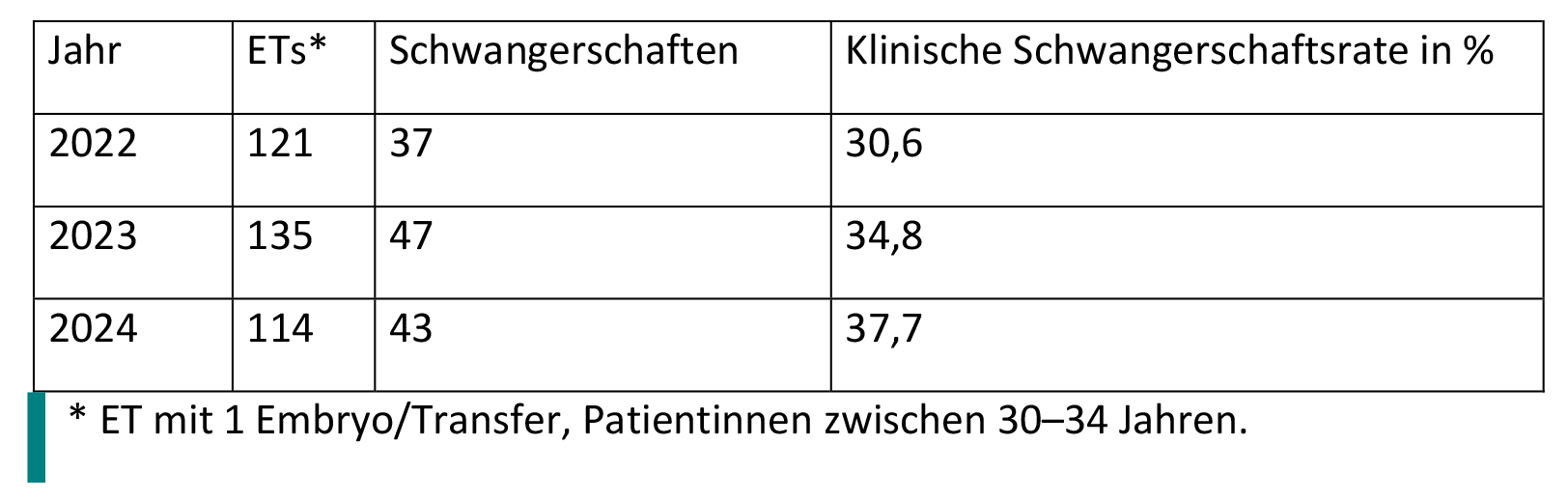
Es wurde in Tab. 1 die klinische Schwangerschaftsrate ausgewertet, nicht die positiven Tests. Pro Transfer wurde immer nur 1 Embryo transferiert.
Fazit
Verschiedene Projekte in diesem Bereich zeigen, dass Reinraumtechnik im ART-Bereich grundsätzlich funktioniert und nicht zwingend zu schlechten Resultaten durch negative Beeinflussung der Keimzellen und Embryonen führen muss, wenn das Ganze sinnvoll und prozessorientiert ausgelegt wird. Dies bedingt definierte Prozesse, ein risikobasiertes Vorgehen sowie ein gelebtes und stimmiges QMS. Dazu braucht es etwas Kreativität und den Mut, bekannte Pfade zu verlassen und die regulatorischen Möglichkeiten und Freiräume optimal zu nutzen. Dafür ist eine frühe Kommunikation mit der zuständigen Landesbehörde unbedingt erforderlich.
Auch gilt es, den Blick für das Gesamtkonzept und die Anwendbarkeit zu behalten – eine Arztpraxis bzw. das Labor eines Spitals ist kein Pharmabetrieb und die vorhandenen Ressourcen sind i. d. R. limitiert. Es bestehen somit in der Realität große Unterschiede in der Gestaltung und technischen Ausführung der Räumlichkeiten von Kinderwunscheinrichtungen. Dies sind i. d. R. Arztpraxen, Fachzentren oder Spitäler bzw. klinische Abteilungen mit dem fachlichen Schwerpunkt Kinderwunsch.
Darüber hinaus handelt es sich am Ende auch nicht um ein steriles Arzneimittel oder einen aseptischen Fertigungsprozess, was primär im Fokus des Annex 1 liegt. Die im ART-Bereich ablaufenden Prozessschritte und das hierfür benötigte Equipment sind keinesfalls vergleichbar mit der klassischen Pharmafertigung und werden durch zu enge Auslegung der Regularien und der damit notwendigen Technik vor allem im Klasse-A-Bereich deutlich erschwert, bzw. sind z. T. sogar nicht mehr durchführbar.
Und trotz aller Risikobetrachtung, sinnvoll eingesetzter Technik, standardisierter Prozesse, qualifiziertem Equipment und geschultem und motiviertem Personal bleibt immer ein Unsicherheitsfaktor im Spiel: nämlich die Tatsache, dass es sich bei den verwendeten Keimzellen und Embryonen immer um individuelles, nicht standardisiertes „Material“ handelt und der Mensch am Ende nicht die Entscheidung trifft über das Gelingen der Bemühungen im Rahmen der Assistierten Reproduktion. Der Erfolg der Bemühungen bei der Assistierten Reproduktion kann somit trotz aller medizinischer und technischer Möglichkeiten nicht garantiert werden.
Der Einsatz von Reinraumtechnik und damit die Schaffung kontrollierter Umgebungsbedingungen, in Verbindung mit einem prozess- und nutzerorientierten QMS führt aber in jedem Fall zu reproduzierbaren Bedingungen, einer schnelleren Erkennung von Fehlern und Abweichungen und einem durchgängig strukturierten und geregelten Vorgehen. Damit ist sichergestellt, dass das Bestmögliche im Sinne der Produkt- und Patientensicherheit unternommen wurde. Trotz anfänglicher Vorbehalte gegenüber den ungewohnten Ansätzen und dem erhöhten Aufwand (zeitlich, materiell, personell und finanziell) überwiegt bei den Nutzern aus verschiedenen Projekten am Ende ein positives Gefühl und Zufriedenheit mit der jeweiligen Umsetzung.
Literatur
[1] Gesetz über den Verkehr mit Arzneimitteln (Arzneimittelgesetz - AMG), in der Fassung der Bekanntmachung vom 12. Dez. 2005 (BGBl. I S. 3394), das zuletzt durch Artikel 3 des Gesetzes vom 17. Dez. 2014 (BGBl. I S. 2222) geändert worden ist
[2] Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) vom 3. Nov. 2006 (BGBl. I S. 2523), die zuletzt durch Artikel 9 des Gesetzes vom 23. Okt. 2024 (BGBl. 2024 I Nr. 324) geändert worden ist
[3] EudraLex - Volume 4 - Good Manufacturing Practice (GMP) guidelines, https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en
[4] Leitfaden der Deutschen Gesellschaft für Chirurgie zur Guten Fachlichen Praxis (GFP) für die Entnahme von menschlichen Geweben und Zellen zur Herstellung eines Arzneimittels, https://www.dgch.de/fileadmin/media/pdf/servicemeldungen/069_Gewebegesetz_GFP-Leitfaden_der_DGCH_fuer_die_Gewinnung_menschlicher_Gewebe.pdf
[5] Leitlinie für die Führung und Einrichtung eines Labors für die Durchführung Assistierter Reproduktionstechnologien beim Menschen (ART-Labor) innerhalb einer reproduktionsmedizinischen Versorgungseinrichtung der Arbeitsgemeinschaft Reproduktionsbiologie des Menschen (AGRBM) vom 21. Mai 2021
[6] Leitlinie zum verantwortlichen Arbeiten im ART-Labor der Arbeitsgemeinschaft Reproduktionsbiologie des Menschen (AGRBM) vom 28. März 2014
[7] ESHRE position paper on the EU Tissues and Cells Directive EC/ 2004/23; Nov. 2007; Air quality: Commission Directive 2006/86/EC, Annex I.D. Facilities/Premises
[8] Gesetz über die Spende, Entnahme und Übertragung von Organen und Geweben (Transplantationsgesetz - TPG) in der Fassung der Bekanntmachung vom 4. Sept. 2007 (BGBl. I S. 2206), das zuletzt durch Artikel 8b des Gesetzes vom 22. März 2024 (BGBl. 2024 I Nr. 101) geändert worden ist"
[9] Gesetz zum Schutz von Embryonen (Embryonenschutzgesetz - ESchG) vom 13. Dez. 1990 (BGBl. I S. 2746), das zuletzt durch Artikel 1 des Gesetzes vom 21. Nov. 2011 (BGBl. I S. 2228) geändert worden ist"
[10] Gesetz zur Verhütung und Bekämpfung von Infektionskrankheiten beim Menschen (Infektionsschutzgesetz - IfSG) vom 20. Juli 2000 (BGBl. I S. 1045), das zuletzt durch Artikel 8v des Gesetzes vom 12. Dez. 2023 (BGBl. 2023 I Nr. 359) geändert worden ist"
[11] ESHRE Revised guidelines for good practice in IVF laboratories; December 2015
[12] Guide to the quality and safety of tissues and cells for human application des European Directorate for the Quality of Medicines & HealthCare (EDQM)
[13] Verordnung (EU) 2024/1938 des Europäischen Parlaments und des Rates vom 13. Juni 2024 über Qualitäts- und Sicherheitsstandards für zur Verwendung beim Menschen bestimmte Substanzen menschlichen Ursprungs und zur Aufhebung der Richtlinien 2002/98/EG und 2004/23/EG
[14] DIN EN ISO 14644-1:2016-06, Reinräume und zugehörige Reinraumbereiche; Deutsche Fassung EN_ISO_14644-1:2015, v. a. Teile 1, 2, 3, 4, 5, 7, 8, 11, 14, 15
[15] VDI 2083 Reinraumtechnik, v. a. Blätter 1, 2, 3, 3.1, 4.1, 5, 6, 8.1, 15, 16.2.
Die Links wurden zuletzt abgerufen am 30. Juni 2025.
Dipl.-Ing. (FH) Claudia Pachl
Frau Pachl ist Ingenieurin für Pharmatechnik und Geschäftsführerin der VALTEC GmbH mit Sitz in Neuhausen am Rheinfall, Schweiz. Sie verfügt über langjährige Erfahrung in der Pharmaindustrie und Medizintechnik. Im Bereich der Assistierten Reproduktionstechnik hat sie bereits einige Zentren und Praxen bei der Erlangung einer Erlaubnis nach § 20 b und c AMG unterstützt. Sie engagiert sich u. a. im Verein Deutscher Ingenieure (VDI) und ist als Autorin, Referentin und Gastdozentin tätig.
![]()
ValTec GmbH
Zentralstrasse 100
8212 Neuhausen
Schweiz
Telefon: +49 173 6983802
Telefax: +41 52 6750001
eMail: claudia.pachl@valtec-gmbh.com
Internet: http://www.valtec-gmbh.com



