



- Science
- Translated with AI
Dipl.-Ing. (FH) Claudia Pachl
Between GMP and Desire for Children
How cleanroom technology can be effectively implemented in assisted reproductive technology
")
Figure 5: Embryo laboratory with ICSI and transfer workstation (Source: Practice for Fertility Dr. Peet and Dr. Wilkening)
")
Figure 4: Egg Cell Laboratory (Source: Practice for Fertility Dr. Peet and Dr. Wilkening)
 and IVF workbenches (Source: ZERM)")
Figure 6: ICSI workstations (as TAV area) and IVF workbenches (Source: ZERM)
")
Figure 7: IVF Workbenches (Source: ZERM)
")
Figure 3: Andrology Laboratory (Source: Fertility Practice Dr. Peet and Dr. Wilkening)
")
Figure 8: Transfer area with passage (Source: ZERM)
")
Figure 9: Andrology Laboratory (Source: ZERM)
")
Figure 10: Material hoist point load (Source: ZERM)
")
Figure 11: Material lock (Source: ZERM)
")
Figure 12: Cryogenic Storage (Source: ZERM)
 Claudia Pachl, VALTEC")
Dipl.-Ing. (FH) Claudia Pachl, VALTEC
Cleanroom Technology in the Field of Assisted Reproductive Technology (ART) is a highly debated topic. Regulated by the Medicines Act (AMG) and the Medicines and Active Substance Manufacturing Regulation (AMWHV), but not comparable to other tissues or even pharmaceutical manufacturing, the end user – fertility clinics and medical practices – are often left puzzled. Cleanroom technology and fertility, at first glance, seem difficult to reconcile. However, there are various options and flexibilities that, when used wisely, can lead to a value-adding and robust system for users and patients.
First published in: cleanroom & processes 4, No. 3, 2–10 (2025)
Cleanroom technology is applied where airborne particles generally have a harmful influence on the product being manufactured and the associated processes. Semiconductor manufacturing, optics and laser technology, aerospace engineering – impossible without cleanrooms and cleanroom technology. But also in the food industry, biotechnology, medical devices, and pharmaceuticals, pure and controlled environments are fundamental – depending on manufacturing processes, product requirements, and application. Besides airborne particles, the microbial count also plays a significant role here.
Without cleanroom technology, there would be no smartphones, no durable sliced cheese, and no (life-saving in emergencies) medications, to name just a few examples. In addition to protecting the user – customer or patient – and the product itself, cleanroom technology also serves to protect the manufacturing personnel and the environment.
Another area where the application of cleanroom technology has increasingly come into focus in recent years is assisted reproductive techniques (ART), often simply referred to as artificial insemination. Since this article does not intend to discuss the various techniques and methods within the broad field of ART – e.g., in-vitro fertilization (IVF), intrauterine insemination (IUI), or intracytoplasmic sperm injection (ICSI) – the term ART will be used broadly here, regardless of the method applied. The focus of this article is to examine the extent to which the use of cleanroom technology in ART provides benefits or is rather harmful. This is a topic of sometimes controversial discussion among users, experts, and authorities.
Legal Foundations
In the ART sector, a multitude of laws and regulations must be considered:
The Medicines Act
Fertility clinics and fertility centers require a permit from the competent state authority according to §§ 20 b and c of the Medicines Act (AMG) [1] to conduct activities within fertility treatments. § 20 b regulates tissue collection and laboratory examinations, while § 20 c covers the processing, conservation, testing, storage, and marketing of tissues or tissue preparations.
It also includes information about the qualified person and their required qualifications, the demand for qualified personnel, as well as suitable rooms and facilities for the intended activities. Furthermore, the AMG mentions the application of Good Professional Practice (GFP) and the consideration of the current state of science and technology during processing, including labeling, conservation, storage, and testing. Lastly, there is a reference to a quality management system (QMS) based on GFP principles, which must be continuously maintained.
Medicines and Active Substance Manufacturing Regulation
Section 5a [2] of the Medicines and Active Substance Manufacturing Regulation (AMWHV) regulates special provisions for tissue collection and tissue donor laboratories. With regard to the focus of this article, § 36 "Processing and storage by the tissue establishment" is particularly relevant. It states in paragraph (2):
"The premises and equipment according to § 5 and the hygiene measures according to § 6 must be suitable to protect the properties of the tissue necessary for its use and to minimize the risk of contamination, especially microbial contamination, during processing:"
"To the extent that tissues are exposed to the environment during processing, this must occur in an environment with defined air quality and cleanliness. The effectiveness of these measures must be validated and monitored."
"If the tissues are not subjected to inactivation or sterilization procedures as per item 1, a clean air grade for microbial and particulate count corresponding to Class A according to the EU-GMP guideline, Annex 1 (Notification of March 12, 2008, BAnz. p. 1217), with a background environment suitable for processing the tissue that, in terms of particles and microbes, at least meets Class D of Annex 1 of the guideline, is required." [2]
This clearly indicates the requirement for a cleanliness class A with a background environment of at least D according to EU-GMP guidelines [3] regarding particles and microbes. The following exceptions are permitted:
"Deviations from the environment requirements are allowed if"
a) a validated procedure for inactivating microbes or terminal sterilization is applied, or
b) it can be demonstrated that exposure to a Class A environment has harmful effects on the properties of the tissue, or
c) it can be demonstrated that, considering the method of use of the tissue in the recipient, there is a significantly lower risk of transmitting bacterial or fungal infections compared to tissue transplantation, or
d) it is technically impossible to perform the required procedure in a Class A environment.
"It must be demonstrated and documented that the chosen environment achieves the necessary quality and safety of the tissue or tissue preparation, at least considering the intended purpose, method of use, and the immune status of the recipient." [2]
Since germ cells in ART cannot be subjected to inactivation or sterilization procedures, the fundamental environmental requirements are clearly defined. The deviations described under c) are already supported by various scientific publications, statements from relevant professional circles, and risk assessments. The transfer of germ cells and embryos to the patient involves various risks but fundamentally differs from tissue transplantation.
As the focus of this article is on the use of cleanroom technology in ART and not on medical details, a detailed discussion is omitted here, and relevant literature and professional publications are referenced.
Options b) and d) are directly related to the application of cleanroom technology and will be examined further in this article.
EU-GMP Guidelines
Annex 1 of the EU-GMP guideline [3] is the basis for the relevant sections in § 36 ff of the AMWHV regarding particles and microbes. The limits defined in Annex 1 are applicable, although the AMWHV still references the old Annex 1 from March 12, 2008. Since the limits for particles and microbes are identical to the current version from August 22, 2022, this does not pose a problem for this discussion.
The other parameters described in Annex 1 for Classes A–D are not mentioned in § 36 ff of the AMWHV and are therefore not explicitly required for the ART sector, e.g., airflow velocity, air exchange, differential pressures, etc.
The EU-GMP guideline, according to the GFP guideline of the German Society for Surgery [4], is not applicable for the collection of human tissues and cells (per § 20 b AMG), provided the collection is performed according to GFP rules.
Working Group for Reproductive Biology of Humans
The Working Group for Reproductive Biology of Humans (AGRBM) has published two guidelines dealing with the operation and setup of an ART laboratory [5] and responsible work in the ART laboratory [6].
These guidelines also address the environmental conditions according to Annex 1 of the EU-GMP guideline and their impact on germ cells. The ART laboratory is considered part of a collection facility according to AGRBM guidelines.
According to AGRBM and the European Society of Human Reproduction and Embryology (ESHRE) [7], the exceptions listed in § 36 of the AMWHV points a) to d) are fully applicable to ART and its processes. Nevertheless, environmental conditions of a cleanliness class D according to Annex 1 are recommended, while class A conditions are considered counterproductive and are not recommended.
Other Regulations and Standards
Besides those already mentioned, numerous other regulations and requirement documents are applicable to ART. Notably, the Transplantation Act [8], Embryo Protection Act [9], Infection Protection Act [10], and ESHRE guidelines [11]. These are supplemented by the European Directorate for the Quality of Medicines & Healthcare (EDQM) Guide [12] and the new Substances of Human Origin (SoHO) Regulation [13], which will be effective from 2027.
Regarding cleanroom technology, relevant parts of the technical rules according to DIN EN 14644 [14] and VDI 2083 [15] must be considered.
Patient Safety
Even without detailed medical knowledge, it is understandable that a transplantation of stem cells or organs differs from artificial fertilization or embryo transfer. In the first case, it usually involves severely ill, sometimes immunosuppressed patients, whose survival depends heavily on tissue transplantation. The transplanted tissue is often specified as sterile, and collection, processing, and handling are usually performed aseptically or under largely aseptic conditions. The process steps and equipment are accordingly adapted, and most processes occur in closed systems. Transplantation typically takes place under surgical conditions.
In ART, recipients are generally healthy or have a good immune status, although there are various reasons for the unfulfilled desire to have children. The collection of germ cells from both women and men cannot be completely germ-free. The reintroduction of processed cells into the uterus of the immunocompetent recipient occurs via a natural body opening, which is naturally colonized with microbes. If external contamination with microbes occurs during extracorporeal cultivation, the cultivation or transfer is at worst suspended. The risk of injury during collection and reintroduction is low and not comparable to transplantation.
Therefore, it can be questioned whether the full application of cleanliness class A for critical steps with a background environment of at least class D is strictly necessary, or whether the safety of germ cells and embryos can also be ensured through microbe-reduced or microbe-free processes in the ART field during processing, thus ultimately safeguarding patient safety.
Cleanroom Technology for ART – Useful or Harmful?
As already mentioned, the regulations only appear to provide clear requirements at first glance because the relevant passages generally refer to tissues and not specifically to ART. The collection, processing, and transfer of tissues are so diverse and case-specific that the regulatory attempt to cover all variants with a common clause is somewhat unfortunate from the author's perspective and can lead to controversy.
For the processing of germ cells in ART falling under § 20 c AMG, inspectors from the authorities expect controlled environmental conditions as required in section 5a of the AMWHV. Additionally, qualification of equipment and devices, monitoring of critical parameters, and a QMS according to GFP are expected. This often leads to discussions and uncertainty among practitioners.
Professional circles emphasize the unrestricted applicability of the exceptions described in the AMWHV (see section on Medicines and Active Substance Manufacturing Regulation) and highlight the harmful influence of cleanroom technology and the associated technical measures and environmental conditions. The AGRBM guideline [5] discusses these exceptions in detail and provides justification.
Particularly, the negative impact of airflow velocity in GMP Class A areas is highlighted, as it can cause cooling effects, drying, changes in extracellular pH, and osmolarity alterations in the medium, ultimately damaging the cells. Furthermore, the technology and hygiene measures required to maintain classical GMP conditions make handling germ cells significantly more difficult, increasing the risk for the product and endangering the germ cells. Some traditional GMP requirements, especially for Class A in ART, are technically difficult or impossible to implement. For example, placing an ICSI microscope with accessories on a workbench is nearly impossible, and performing the corresponding process steps correctly is almost unfeasible.
Inspecting authorities are bound by the applicable regulations and consequently require appropriate evidence and documentation to demonstrate compliance. Statements from working groups and professional circles are often overlooked. This challenging situation can be resolved with some creativity.
Solution Approaches
The author suspects that, most likely due to a lack of alternatives, § 5a of the AMWHV refers to Annex 1 of the EU-GMP guideline [3] regarding the definition of particles and microbes.
Upon closer examination, this reference is limited exclusively to these parameters. All other relevant parameters described in Annex 1, such as differential pressures, air exchange, recovery times, etc., are not mentioned or required. The airflow velocity, which is implemented as a turbulence-free displacement airflow with 0.36 m/s to 0.52 m/s for Class A as the state of the art, is not explicitly mandated in the applicable sections for tissues – and thus also for ART. This leaves room for interpretation and creative implementation, offering various possibilities for technical realization that would not be feasible in classical pharmaceutical manufacturing or medical technology.
Moreover, the EU-GMP guideline, originally created as a directive for the manufacture of medicines for humans and animals under defined conditions with standardized procedures and reproducible results, explicitly focuses on sterile pharmaceutical production in Annex 1.
Processes in ART follow method-dependent, established procedures. Nonetheless, even with identical environmental conditions and process structures, highly variable results are expected on a patient-specific basis. For example, oocytes from the same donor can have different qualities, develop differently under the same conditions and media, and be more or less suitable for further use. The final product is by no means sterile.
From the author's perspective, Annex 1 of the EU-GMP guideline can serve as a guideline to meet particle and microbial count requirements. However, the full implementation of the other requirements of Annex 1 is not explicitly mandated for ART. The particle and microbial counts according to Annex 1 are required, but without the technical effort currently necessary in (cleanroom) technology.
What might this look like in practice? Unfortunately, it cannot be achieved without technical equipment and installations. To reliably ensure particles and microbes of class D or A, certain conditions are necessary. The air must be properly directed and processed, critical parameters monitored, surfaces suitable regarding particle emission, volatile organic compounds (VOCs), and resistance to cleaning and disinfecting agents.
This also requires certain structural prerequisites to avoid issues elsewhere, e.g., with occupational safety (e.g., ceiling ducts). Such technical installations in buildings designed as practices or offices are challenging or sometimes impossible to implement.
Volatile Organic Compounds
At this point, another significant aspect should be mentioned, which is not addressed in the regulations of inspecting authorities: VOCs. These are gaseous and vaporous organic substances in the air, such as hydrocarbons, alcohols, aldehydes, and organic acids. They can originate from materials used in surfaces (ceilings, floors, walls, furniture, etc.) or be generated during cleaning and disinfection.
VOCs are particularly harmful to germ cells in ART and should therefore be minimized or avoided. They have a cytotoxic effect and can negatively influence the quality and development of germ cells. Numerous publications from relevant professional circles confirm this.
Paradoxically, this aspect is rarely considered in authority inspections because the relevant regulations and guidelines for approval under AMG §§ 20 b and c [1] do not include specific requirements regarding VOCs. However, it must be considered in the planning and design of rooms and environmental conditions in ART. This poses a particular challenge for cleaning and disinfection to ensure compatibility with the established GMP requirements for germ cells.
The negative impact of VOCs on germ cells is significantly greater than potential contamination by particles and microbes. Guidance on VOCs can be found in DIN EN ISO 14644-8 (classification of air cleanliness based on chemical concentration) [15] and VDI 2083-8.1 (air cleanliness based on chemical concentration, ACC) [16]. However, specific limits for ART are not available in the literature.
Practical Examples
Below are two project examples illustrating how compliance can be achieved between regulatory requirements for permits under AMG §§ 20 b and c, GFP requirements, and professional standards, as well as process-specific, operator-dependent conditions.
Case 1:
Fertility practice Dr. Peet and Dr. Wilkening in Berlin, in a building complex with a hotel and other commercial spaces. The practice moved into new rooms in 2016. Initial situation (Fig. 1):
– No clear separation of the ART area
– No coordinated personnel and material flow concept
– No controlled environmental conditions
– No qualification of equipment and environment

Figure 1: Layout of the ART area. (Source: Fertility Practice Dr. Peet and Dr. Wilkening)
The responsible authority denied the permit under AMG §§ 20 b and c. The approach, process flows, and environmental conditions were similar to the previous rooms on the same street. The move to new rooms resulted in the loss of grandfather rights, and full compliance with the requirements of the AMG [1] and AMWHV [2] was necessary.
Implementation:
The layout regarding room geometry was not changed. However, the laboratory area was clearly separated as a cleanroom from other practice areas, and the access concept was adjusted accordingly. Cleanroom classes were defined based on Annex 1 of the EU-GMP guideline, with rooms and traffic areas mainly corresponding to Class D (Fig. 2, shown in red). The handling of germ cells occurs under Class A conditions (Fig. 2, shown in blue), mainly achieved through areas with turbulence-free displacement airflow (TAV). It is particularly notable that this is an open A-in-D concept. This was necessary to ensure ART-specific process flows with minimal restrictions while maximizing microbial reduction (Figs. 3, 4, and 5). Despite process-relevant equipment in the A-area – e.g., centrifuges, incubators, microscopes with warming plates – turbulence-free displacement airflow is achieved with reduced airflow velocity. This is verified through regular measurements to demonstrate qualified conditions according to DIN EN ISO 14644 [14].

Figure 2: GMP-compliant layout of the ART area. (Source: Fertility Practice Dr. Peet and Dr. Wilkening)
Case 2:
IVF laboratory at the Center for Endocrinology and Reproductive Medicine (ZERM) at the University Hospital Würzburg
Initial situation:
– Existing rooms outdated and too small
– Renovation in the tower over 3 floors, IVF lab on the 3rd floor
– Building gutted and newly planned
Implementation:
In this project, a completely new layout concept for the laboratory was developed in the gutted floor. Again, a Class A in D concept was used. Due to specific customer requirements, the Class A areas are realized either via IVF workbenches (Figs. 6, 7, and 9) or a large TAV area with two ICSI workstations (Fig. 6).
A particular challenge was transporting the oocytes from the 1st floor to the IVF laboratory on the 3rd floor. For this, a GMP-compliant material lift (Fig. 10) was constructed, ensuring conditions of Class D inside and providing the required pressure cascade or airflow direction from the IVF lab to the collection room across two floors.
Additionally, a pass-through (Fig. 8) was installed next to the andrology laboratory leading to the adjacent transfer room (outside the cleanroom area), through which transfer catheters are passed from the IVF lab into the transfer room and back. The cryo-storage (Fig. 12) is accessible via the black area of the material lock and integrated into the overall access concept. As a CNC area (controlled, not classified), it has less strict environmental requirements but fits well into the process flow and hygiene concept.
Solution approaches in both projects:
- Review and adjustment of processes and workflows to the new conditions
- Risk-based identification of critical process steps and parameters
- Clear delineation of the pure area, including access concept
- Securing critical process steps and parameters (technical, organizational)
- Creating optimal environmental conditions without negatively affecting germ cells
- Optimal arrangement of areas and installations
- Installation of HVAC systems and areas with turbulence-free displacement airflow (Fig. 6) and IVF workbenches (Figs. 7, 9) with adjusted parameters
- Adjustment of the hygiene concept (clothing, cleaning, disinfection)
- Qualification of all relevant devices, environmental conditions, storage
- Proof of effectiveness of cleaning and disinfection
- QMS, including Standard Operating Procedures (SOPs)
Through targeted technical measures, such as HVAC systems, process-appropriate dimensioned TAV areas, IVF workbenches (where required), pressure cascade concepts, suitable surfaces and materials (keyword: VOC), and user-friendly hygiene and clothing concepts, optimal conditions were created to enable safe handling of germ cells with minimal interference in workflows from cleanroom technology. Especially, creating large TAV areas as open Class A zones within a Class D environment generally allows much better handling during process steps than would be possible on a workbench. Of course, this requires appropriate building technical conditions.
The concept was further complemented by application-appropriate and sensible parameter settings, e.g., reducing airflow velocity in the A-areas to well below 0.36 m/s to minimize negative effects on germ cells during open handling. The maintenance of turbulence-free displacement airflow despite low velocity and installed equipment (e.g., incubators, microscopes with warming plates, centrifuges) was reliably demonstrated through airflow visualization.
The open A-in-D concept ensures compliance with the regulatory minimum requirements of the AMWHV. The particle and microbial limits according to Annex 1 of the EU-GMP guideline are reliably met (values have been monitored in the Fertility Practice since 2017 and in the UKW since late 2023). In other life sciences areas, this approach might be considered non-discussable, but in IVF, it is feasible and proven in practice. This approach was successfully implemented in a hospital stem cell laboratory in another project as well.
Through a correspondingly designed continuous monitoring system in ART, installed and parameterized based on risk, critical parameters are permanently monitored. No product-related particle monitoring takes place in Class A, as this was excluded through risk assessment. Hygiene monitoring and intermittent monitoring to maintain qualified conditions complete the surveillance concept.
All relevant devices, equipment, and environmental conditions were fully qualified. The effectiveness of cleaning and disinfection agents was demonstrated.
The basis for all work is an adapted QMS, modified accordingly for this area. Since users in an IVF laboratory generally do not have resources comparable to those in the pharmaceutical industry, the QMS must consider regulatory requirements as well as product and patient safety, but also be manageable in effort to ensure long-term adherence.
Statistics
For the IVF laboratory at UKW, the following brief statistics were provided by the user (Tab. 1):
((Repro width 82mm))
Table 1: Statistics on embryo transfers (ETs) and pregnancies.
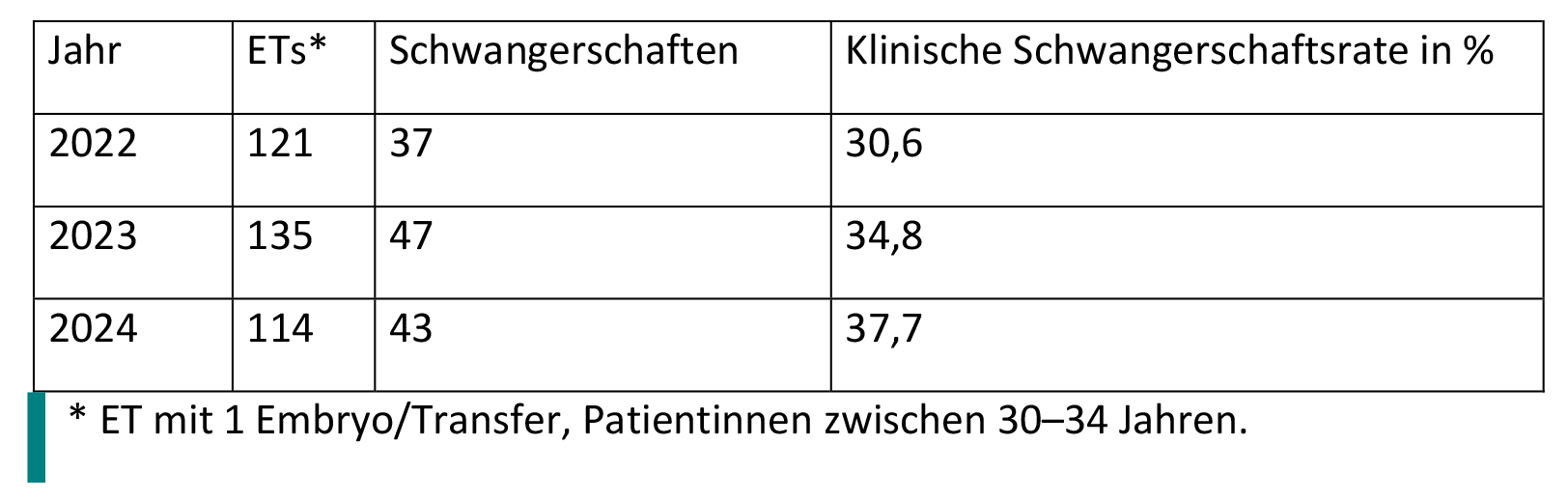
Table 1 shows the clinical pregnancy rate, not positive tests. Only one embryo was transferred per cycle.
Conclusion
Various projects in this area demonstrate that cleanroom technology in ART generally works and does not necessarily lead to poor results due to negative influence on germ cells and embryos when designed sensibly and process-oriented. This requires defined processes, a risk-based approach, and a well-maintained and consistent QMS. It also requires some creativity and the courage to leave familiar paths and to make optimal use of regulatory possibilities and freedoms. Early communication with the responsible authorities is essential for this.
It is also important to keep an overall view and consider applicability – a medical practice or a hospital laboratory is not a pharmaceutical operation, and resources are usually limited. There are significant differences in the design and technical implementation of fertility clinics, typically practices, specialized centers, or hospital departments with a focus on fertility.
Furthermore, the process steps and equipment used in ART are not comparable to sterile pharmaceutical production, which is the primary focus of Annex 1. The process steps and equipment in ART are not comparable to traditional pharmaceutical manufacturing and are significantly hindered or even impossible to implement due to overly strict regulations and technical requirements, especially in Class A environments.
Despite all risk considerations, the use of appropriate technology, standardized processes, qualified equipment, and trained personnel, there remains an inherent uncertainty: germ cells and embryos are always individual, non-standardized "materials," and ultimately, humans do not decide whether the attempts at assisted reproduction succeed. The success cannot be guaranteed despite all medical and technical possibilities.
However, the use of cleanroom technology and the creation of controlled environments, combined with a process- and user-oriented QMS, lead in any case to reproducible conditions, faster detection of errors and deviations, and a consistently structured and regulated approach. This ensures that the best possible measures are taken in the interest of product and patient safety. Despite initial reservations against unfamiliar approaches and increased effort (time, material, personnel, and financial), users from various projects ultimately feel positive and satisfied with the implementation.
References
[1] Medicines Act (Arzneimittelgesetz - AMG), as published on December 12, 2005 (BGBl. I p. 3394), last amended by Article 3 of the Act of December 17, 2014 (BGBl. I p. 2222)
[2] Medicines and Active Substance Manufacturing Regulation (AMWHV) of November 3, 2006 (BGBl. I p. 2523), last amended by Article 9 of the Act of October 23, 2024 (BGBl. 2024 I No. 324)
[3] EudraLex - Volume 4 - Good Manufacturing Practice (GMP) guidelines, https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en
[4] Guideline of the German Society for Surgery on Good Professional Practice (GFP) for the collection of human tissues and cells for medicinal product manufacturing, https://www.dgch.de/fileadmin/media/pdf/servicemeldungen/069_Gewebegesetz_GFP-Leitfaden_der_DGCH_fuer_die_Gewinnung_menschlicher_Gewebe.pdf
[5] Guideline for the operation and setup of a laboratory for assisted reproductive technologies in humans (ART-Labor) within a reproductive medicine facility of the Working Group for Reproductive Biology of Humans (AGRBM), May 21, 2021
[6] Guideline on responsible work in the ART laboratory of the Working Group for Reproductive Biology of Humans (AGRBM), March 28, 2014
[7] ESHRE position paper on the EU Tissues and Cells Directive EC/2004/23; Nov. 2007; Air quality: Commission Directive 2006/86/EC, Annex I.D. Facilities/Premises
[8] Transplantation Act (Transplantationsgesetz - TPG) in the version published on September 4, 2007 (BGBl. I p. 2206), last amended by Article 8b of the Act of March 22, 2024 (BGBl. 2024 I No. 101)
[9] Embryo Protection Act (Embryonenschutzgesetz - ESchG) of December 13, 1990 (BGBl. I p. 2746), last amended by Article 1 of the Act of November 21, 2011 (BGBl. I p. 2228)
[10] Infection Protection Act (Infektionsschutzgesetz - IfSG) of July 20, 2000 (BGBl. I p. 1045), last amended by Article 8v of the Act of December 12, 2023 (BGBl. 2023 I No. 359)
[11] ESHRE revised guidelines for good practice in IVF laboratories; December 2015
[12] Guide to the quality and safety of tissues and cells for human application by the European Directorate for the Quality of Medicines & Healthcare (EDQM)
[13] Regulation (EU) 2024/1938 of the European Parliament and the Council of June 13, 2024, on quality and safety standards for substances of human origin intended for human use and repealing Directives 2002/98/EC and 2004/23/EC
[14] DIN EN ISO 14644-1:2016-06, Cleanrooms and associated controlled environments; German version EN ISO 14644-1:2015, mainly parts 1, 2, 3, 4, 5, 7, 8, 11, 14, 15
[15] VDI 2083 Cleanroom technology, mainly sheets 1, 2, 3, 3.1, 4.1, 5, 6, 8.1, 15, 16.2.
The links were last accessed on June 30, 2025.
Dipl.-Ing. (FH) Claudia Pachl
Ms. Pachl is an engineer for pharmaceutical technology and managing director of VALTEC GmbH, based in Neuhausen am Rheinfall, Switzerland. She has many years of experience in the pharmaceutical industry and medical technology. In the field of assisted reproductive technology, she has already supported several centers and practices in obtaining permits under §§ 20 b and c AMG. She is involved, among other things, in the German Engineers Association (VDI) and works as an author, speaker, and guest lecturer.
![]()
ValTec GmbH
Zentralstrasse 100
8212 Neuhausen
Switzerland
Phone: +49 173 6983802
Fax: +41 52 6750001
email: claudia.pachl@valtec-gmbh.com
Internet: http://www.valtec-gmbh.com



